使用 Bio.Phylo 进行系统发育分析
Bio.Phylo 模块在 Biopython 1.54 中引入。它遵循 SeqIO 和 AlignIO 的思路,旨在提供一种通用的方法来处理系统发育树,与源数据格式无关,并提供一致的 I/O 操作 API。
Bio.Phylo 在一篇开放获取的期刊文章 Talevich *等人* 2012 [Talevich2012] 中有介绍,你可能也会发现这篇论文很有帮助。
演示:树中有什么?
为了熟悉该模块,我们从一个已经构建好的树开始,并以几种不同的方式检查它。然后,我们将对分支进行着色,以使用 phyloXML 的特殊功能,最后保存它。
使用你喜欢的文本编辑器创建一个名为 simple.dnd 的简单 Newick 文件,或者使用 Biopython 源代码提供的 simple.dnd
(((A,B),(C,D)),(E,F,G));
这棵树没有分支长度,只有拓扑结构和标记的终端。(如果你有一个实际的树文件,你可以使用它来执行此演示。)
启动你选择的 Python 解释器
$ ipython -pylab
对于交互式工作,使用 -pylab 标志启动 IPython 解释器可以启用 **matplotlib** 集成,这样图形会自动弹出。我们将在本演示中使用它。
现在,在 Python 中,读取树文件,提供文件名和格式名称。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
将树对象打印为字符串可以让我们看到整个对象层次结构。
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade()
Clade()
Clade()
Clade(name='A')
Clade(name='B')
Clade()
Clade(name='C')
Clade(name='D')
Clade()
Clade(name='E')
Clade(name='F')
Clade(name='G')
Tree 对象包含有关树的全局信息,例如它是有根的还是无根的。它有一个根枝,在它之下,它嵌套了从根到顶端的枝列表。
draw_ascii 函数创建一个简单的 ASCII 艺术(纯文本)树状图。这是一种方便的交互式探索可视化方式,以防没有更好的图形工具可用。
>>> from Bio import Phylo
>>> tree = Phylo.read("simple.dnd", "newick")
>>> Phylo.draw_ascii(tree)
________________________ A
________________________|
| |________________________ B
________________________|
| | ________________________ C
| |________________________|
_| |________________________ D
|
| ________________________ E
| |
|________________________|________________________ F
|
|________________________ G
如果你安装了 **matplotlib** 或 **pylab**,你可以使用 draw 函数创建一个图形树。
>>> tree.rooted = True
>>> Phylo.draw(tree)
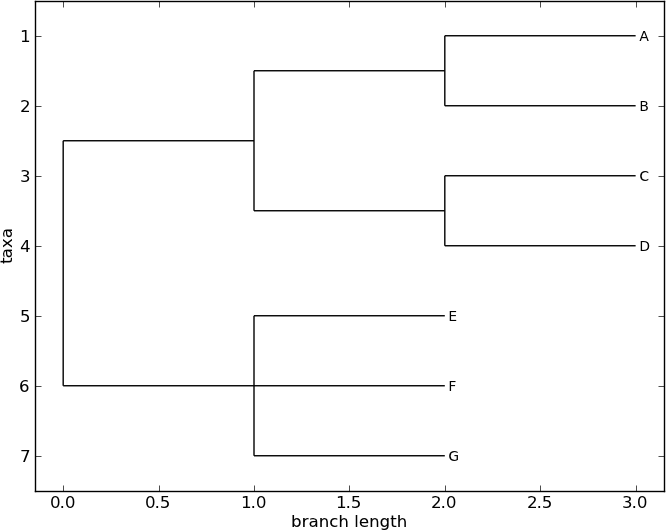
图 6 使用 Phylo.draw 绘制的带根树。
参见 图 6。
在树中对分支进行着色
draw 函数支持在树中显示不同的颜色和分支宽度。从 Biopython 1.59 开始, color 和 width 属性在基本 Clade 对象上可用,使用它们不需要额外的操作。这两个属性都引用到给定枝的引导枝,并递归地应用,因此所有后代枝在显示过程中也将继承分配的宽度和颜色值。
在早期版本的 Biopython 中,这些是 PhyloXML 树的特殊功能,使用这些属性需要先将树转换为基本树对象的子类,称为 Phylogeny,来自 Bio.Phylo.PhyloXML 模块。
在 Biopython 1.55 及更高版本中,这是一个方便的树方法
>>> tree = tree.as_phyloxml()
在 Biopython 1.54 中,你可以通过额外导入一个模块来实现相同的功能
>>> from Bio.Phylo.PhyloXML import Phylogeny
>>> tree = Phylogeny.from_tree(tree)
请注意,Newick 和 Nexus 格式不支持分支颜色或宽度,因此如果你在 Bio.Phylo 中使用这些属性,你只能将这些值保存为 PhyloXML 格式。(你仍然可以将树保存为 Newick 或 Nexus,但颜色和宽度值将在输出文件中被跳过。)
现在我们可以开始分配颜色了。首先,我们将根枝的颜色设置为灰色。我们可以通过将 24 位颜色值分配为 RGB 三元组、HTML 风格的十六进制字符串或预定义颜色之一的名称来做到这一点。
>>> tree.root.color = (128, 128, 128)
或者
>>> tree.root.color = "#808080"
或者
>>> tree.root.color = "gray"
枝的颜色被视为级联到整个枝,因此当我们在这里给根枝着色时,它会将整个树变成灰色。我们可以通过在树的更下方分配不同的颜色来覆盖它。
让我们以“E”和“F”节点的最近共同祖先(MRCA)为目标。 common_ancestor 方法返回原始树中该枝的引用,因此当我们给该枝着色为“salmon”时,颜色会显示在原始树中。
>>> mrca = tree.common_ancestor({"name": "E"}, {"name": "F"})
>>> mrca.color = "salmon"
如果我们碰巧知道某个枝在树中的确切位置,即嵌套列表条目,我们可以通过索引直接跳转到树中的那个位置。这里,索引 [0,1] 指的是根的第一个子节点的第二个子节点。
>>> tree.clade[0, 1].color = "blue"
最后,展示我们的工作
>>> Phylo.draw(tree)
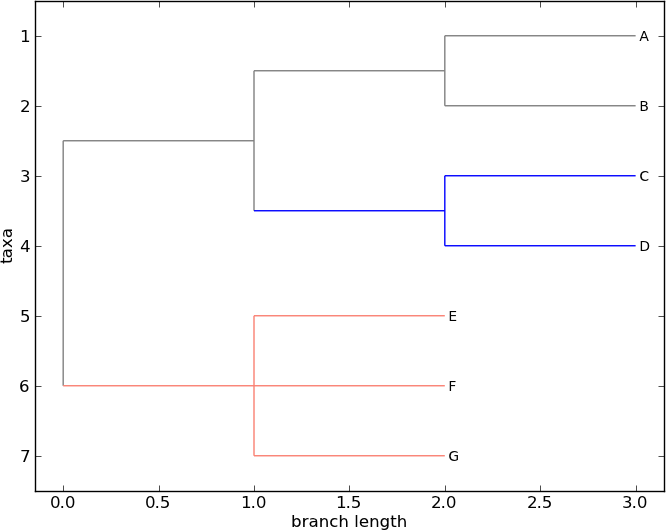
图 7 使用 Phylo.draw 绘制的彩色树。
参见 图 7。
请注意,枝的颜色包括引导到该枝的分支,以及它的后代。E 和 F 的共同祖先恰好在根的下方,通过这种着色,我们可以准确地看到树的根在哪里。
我的,我们已经取得了很大成就!让我们在这里休息一下,保存我们的工作。用文件名或句柄调用 write 函数——这里我们使用标准输出,以查看将要写入的内容——以及格式 phyloxml。PhyloXML 保存了我们分配的颜色,因此你可以在另一个树查看器(如 Archaeopteryx)中打开此 phyloXML 文件,颜色也会在那里显示。
>>> import sys
>>> n = Phylo.write(tree, sys.stdout, "phyloxml")
<phyloxml ...>
<phylogeny rooted="true">
<clade>
<color>
<red>128</red>
<green>128</green>
<blue>128</blue>
</color>
<clade>
<clade>
<clade>
<name>A</name>
</clade>
<clade>
<name>B</name>
</clade>
</clade>
<clade>
<color>
<red>0</red>
<green>0</green>
<blue>255</blue>
</color>
<clade>
<name>C</name>
</clade>
...
</clade>
</phylogeny>
</phyloxml>
>>> n
1
本章的其余部分将更详细地介绍 Bio.Phylo 的核心功能。有关使用 Bio.Phylo 的更多示例,请参阅 Biopython.org 上的食谱页面
I/O 功能
与 SeqIO 和 AlignIO 一样,Phylo 通过四个函数处理文件输入和输出: parse、read、write 和 convert,它们都支持树文件格式 Newick、NEXUS、phyloXML 和 NeXML,以及比较数据分析本体(CDAO)。
read 函数解析给定文件中的单个树并返回它。注意;如果文件包含多棵树或没有树,它将引发错误。
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Nexus/int_node_labels.nwk", "newick")
>>> print(tree)
Tree(rooted=False, weight=1.0)
Clade(branch_length=75.0, name='gymnosperm')
Clade(branch_length=25.0, name='Coniferales')
Clade(branch_length=25.0)
Clade(branch_length=10.0, name='Tax+nonSci')
Clade(branch_length=90.0, name='Taxaceae')
Clade(branch_length=125.0, name='Cephalotaxus')
...
(示例文件可以在 Biopython 发行版中 Tests/Nexus/ 和 Tests/PhyloXML/ 目录中找到。)
要处理多个(或未知数量的)树,请使用 parse 函数迭代给定文件中的每棵树
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> for tree in trees:
... print(tree)
...
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
...
使用 write 函数将树或树的可迭代对象写回文件
>>> trees = Phylo.parse("Tests/PhyloXML/phyloxml_examples.xml", "phyloxml")
>>> tree1 = next(trees)
>>> Phylo.write(tree1, "tree1.nwk", "newick")
1
>>> Phylo.write(trees, "other_trees.xml", "phyloxml") # write the remaining trees
12
使用 convert 函数在任何支持的格式之间转换文件
>>> Phylo.convert("tree1.nwk", "newick", "tree1.xml", "nexml")
1
>>> Phylo.convert("other_trees.xml", "phyloxml", "other_trees.nex", "nexus")
12
要使用字符串作为输入或输出而不是实际文件,请使用 StringIO,就像你在 SeqIO 和 AlignIO 中一样
>>> from Bio import Phylo
>>> from io import StringIO
>>> handle = StringIO("(((A,B),(C,D)),(E,F,G));")
>>> tree = Phylo.read(handle, "newick")
查看和导出树
查看 Tree 对象最简单的方法是 print 它
>>> from Bio import Phylo
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> print(tree)
Phylogeny(description='phyloXML allows to use either a "branch_length" attribute...', name='example from Prof. Joe Felsenstein's book "Inferring Phyl...', rooted=True)
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
Clade(branch_length=0.23, name='B')
Clade(branch_length=0.4, name='C')
这本质上是 Biopython 用于表示树的对象层次结构的提纲。但更可能的是,你希望看到树的图形。有三个函数可以做到这一点。
正如我们在演示中看到的, draw_ascii 将树的 ASCII 艺术图形(带根的系统发育树)打印到标准输出,或者如果给出,打印到打开的文件句柄。并非所有有关树的可用信息都会显示,但它提供了一种无需依赖任何外部依赖项即可快速查看树的方法。
>>> tree = Phylo.read("PhyloXML/example.xml", "phyloxml")
>>> Phylo.draw_ascii(tree)
__________________ A
__________|
_| |___________________________________________ B
|
|___________________________________________________________________________ C
draw 函数使用 matplotlib 库绘制更具吸引力的图像。有关它接受的参数以自定义输出的详细信息,请参阅 API 文档。
>>> Phylo.draw(tree, branch_labels=lambda c: c.branch_length)
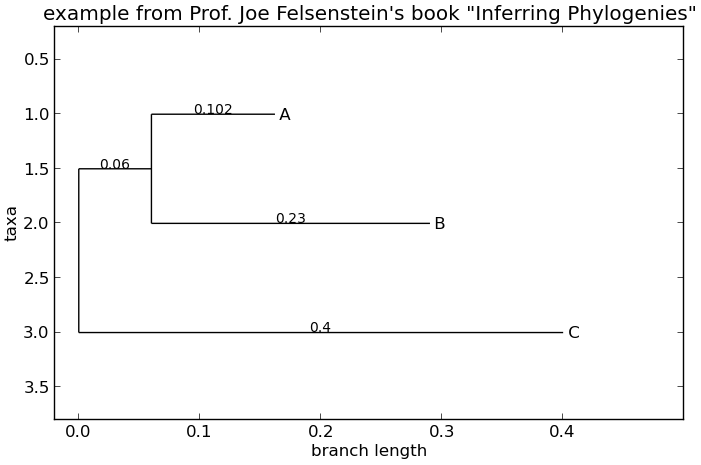
图 8 使用 draw 函数绘制的简单带根树。
参见 图 8 作为示例。
请参阅 Biopython wiki 上的 Phylo 页面 (https://biopython.pythonlang.cn/wiki/Phylo),以了解有关 draw_ascii、draw_graphviz 和 to_networkx 中更高级功能的描述和示例。
使用 Tree 和 Clade 对象
parse 和 read 生成的 Tree 对象是递归子树的容器,附加到 Tree 对象的 root 属性(无论系统发育树是否被认为是生根的)。Tree 具有针对系统发育的全局应用信息,例如生根性,以及对单个 Clade 的引用;Clade 具有节点和进化枝特定的信息,例如分支长度,以及其自身后代 Clade 实例的列表,附加在 clades 属性上。
因此,tree 和 tree.root 之间存在区别。然而,在实践中,您很少需要担心这个问题。为了消除这种差异,Tree 和 Clade 都继承自 TreeMixin,后者包含用于通常用于搜索、检查或修改树或其任何进化枝的方法的实现。这意味着 tree 支持的大多数方法也适用于 tree.root 及其下面的任何进化枝。(Clade 也具有 root 属性,该属性返回进化枝对象本身。)
搜索和遍历方法
为了方便起见,我们提供了一些简化的方法,这些方法直接将所有外部或内部节点作为列表返回
get_terminals创建一个包含此树的所有末端(叶)节点的列表。
get_nonterminals创建一个包含此树的所有非末端(内部)节点的列表。
这些都包装了一个对树遍历具有完全控制的方法,find_clades。另外两个遍历方法,find_elements 和 find_any,依赖于相同核心功能并接受相同的参数,我们将其称为“目标规范”,因为没有更好的描述。这些指定在迭代期间将匹配和返回树中的哪些对象。第一个参数可以是以下任何类型
一个 **TreeElement 实例**,树元素将通过身份匹配 - 因此使用 Clade 实例作为目标进行搜索将在树中找到该进化枝;
一个 **字符串**,匹配树元素的字符串表示 - 特别是进化枝的
name(在 Biopython 1.56 中添加);一个 **类** 或 **类型**,其中相同类型(或子类型)的每个树元素都将匹配;
一个 **字典**,其中键是树元素属性,而值与每个树元素的对应属性匹配。这一个变得更加详细
如果给出
int,则匹配数值相等的属性,例如 1 将匹配 1 或 1.0如果给出布尔值(True 或 False),则将相应属性值评估为布尔值,并检查是否相同
None匹配None如果给出字符串,则该值将被视为正则表达式(该表达式必须匹配相应元素属性中的整个字符串,而不仅仅是前缀)。没有特殊正则表达式字符的给定字符串将完全匹配字符串属性,因此,如果您不使用正则表达式,请不要担心。例如,在进化枝名称为 Foo1、Foo2 和 Foo3 的树中,
tree.find_clades({"name": "Foo1"})匹配 Foo1,{"name": "Foo.*"}匹配所有三个进化枝,而{"name": "Foo"}不会匹配任何内容。
由于浮点运算可能产生一些奇怪的行为,我们不支持直接匹配
float。相反,使用布尔值True匹配指定属性中值为非零的每个元素,然后使用不等式(或精确数字,如果您喜欢冒险生活)手动过滤该属性。如果字典包含多个条目,则匹配元素必须匹配每个给定的属性值 - 考虑“并且”,而不是“或者”。
一个 **函数**,它接受单个参数(它将应用于树中的每个元素),并返回 True 或 False。为了方便起见,LookupError、AttributeError 和 ValueError 被静默,因此这提供了一种安全的方式来搜索树中的浮点值,或一些更复杂的特征。
在目标之后,有两个可选关键字参数
- terminal
— 一个布尔值,用于选择或反对末端进化枝(也称为叶节点):True 搜索仅末端进化枝,False 搜索非末端(内部)进化枝,默认值 None 搜索末端和非末端进化枝,以及任何缺少
is_terminal方法的树元素。- order
— 树遍历顺序:
"preorder"(默认)是深度优先搜索,"postorder"是子节点在父节点之前的 DFS,而"level"是广度优先搜索。
最后,这些方法接受任意关键字参数,这些参数的处理方式与字典目标规范相同:键指示要搜索的元素属性的名称,而参数值(字符串、整数、None 或布尔值)与找到的每个属性的值进行比较。如果未给出任何关键字参数,则匹配任何 TreeElement 类型。此代码通常比将字典作为目标规范传递更短:tree.find_clades({"name": "Foo1"}) 可以缩短为 tree.find_clades(name="Foo1")。
(在 Biopython 1.56 或更高版本中,这甚至可以更短:tree.find_clades("Foo1"))
现在我们已经掌握了目标规范,以下是用作遍历树的方法
find_clades查找包含匹配元素的每个进化枝。也就是说,与
find_elements一样查找每个元素,但返回相应的进化枝对象。(这通常是您想要的。)结果是所有匹配对象的迭代器,默认情况下以深度优先方式搜索。这并不一定与元素在新奇、Nexus 或 XML 源文件中的出现顺序相同!
find_elements查找匹配给定属性的所有树元素,并返回匹配的元素本身。简单的 Newick 树没有复杂的子元素,因此在这些树上,此方法的行为与
find_clades相同。PhyloXML 树通常确实具有附加到进化枝的复杂对象,因此此方法对于提取这些对象很有用。find_any返回
find_elements()找到的第一个元素,或 None。这对于检查树中是否存在任何匹配元素也很有用,并且可以在条件中使用。
另外两个方法有助于在树中的节点之间导航
get_path列出树根(或当前进化枝)与给定目标之间直接的进化枝。返回沿此路径的所有进化枝对象的列表,以给定目标结束,但不包括根进化枝。
trace列出此树中两个目标之间的所有进化枝对象。不包括开始,包括结束。
信息方法
这些方法提供有关整棵树(或任何进化枝)的信息。
common_ancestor查找所有给定目标的最近共同祖先。(这将是一个 Clade 对象)。如果没有给出目标,则返回当前进化枝(调用此方法的进化枝)的根;如果给出 1 个目标,则返回目标本身。但是,如果任何指定的 target 未在当前树(或进化枝)中找到,则会引发异常。
count_terminals计算树中末端(叶)节点的数量。
depths创建树进化枝到深度的映射。结果是一个字典,其中键是树中的所有 Clade 实例,而值是从根到每个进化枝的距离(包括末端)。默认情况下,距离是通向进化枝的累积分支长度,但使用
unit_branch_lengths=True选项时,仅计算分支数量(树中的级别)。distance计算两个目标之间分支长度的总和。如果只指定了一个目标,则另一个目标是此树的根。
total_branch_length计算此树中所有分支长度的总和。这通常被称为系统发育中的“长度”,但我们使用更明确的名称来避免与 Python 术语混淆。
其余这些方法是布尔检查
is_bifurcating如果树是严格二叉的,则为 True;即所有节点都有 2 或 0 个子节点(分别为内部或外部)。根可以有 3 个后代,仍然被认为是二叉树的一部分。
is_monophyletic测试所有给定目标是否构成完整的子进化枝 - 即,是否存在一个进化枝,其末端与给定目标相同。目标应该是树的末端。为了方便起见,如果目标是单系的,则此方法返回目标的共同祖先(MCRA)(而不是值
True),否则返回False。is_parent_of如果目标是此树的后代,则为 True - 不需要是直接后代。要检查进化枝的直接后代,只需使用列表成员资格测试:
if subclade in clade: ...is_preterminal如果所有直接后代都是末端,则为 True;如果任何直接后代不是末端,则为 False。
修改方法
这些方法对树进行就地修改。如果您想保持原始树完整,请首先使用 Python 的 copy 模块创建树的完整副本
tree = Phylo.read("example.xml", "phyloxml")
import copy
newtree = copy.deepcopy(tree)
collapse从树中删除目标,将它的子节点重新链接到它的父节点。
collapse_all折叠此树的所有后代,只留下末端。分支长度得以保留,即到每个末端的距离保持不变。使用目标规范(见上文),仅折叠匹配规范的内部节点。
ladderize根据末端节点的数量对进化枝进行就地排序。默认情况下,最深的进化枝放在最后。使用
reverse=True将进化枝从最深到最浅排序。prune从树中修剪一个终端分支。如果分类单元来自一个分叉,连接节点将被折叠,其分支长度将被添加到剩余的终端节点。这可能不再是一个有意义的值。
root_with_outgroup用包含给定目标的外群分支重新根植这棵树,即外群的共同祖先。此方法仅适用于 Tree 对象,不适用于 Clade 对象。
如果外群与 self.root 相同,则不会发生任何变化。如果外群分支是终端的(例如,给定一个单一的终端节点作为外群),则会创建一个新的分叉根分支,它有一个 0 长度的分支到给定的外群。否则,外群基部的内部节点将成为整棵树的三叉根。如果原始根是分叉的,它将从树中删除。
在所有情况下,树的总分支长度保持不变。
root_at_midpoint将这棵树重新根植在树中两个最远端点之间的计算中点处。(这在内部使用
root_with_outgroup。)split生成 *n*(默认值为 2)个新后代。在物种树中,这是一个物种形成事件。新分支具有给定的
branch_length,并且与该分支的根具有相同的名称,外加一个整数后缀(从 0 开始计数)——例如,分割一个名为“A”的分支会产生“A0”和“A1”这两个子分支。
有关使用可用方法的更多示例,请参阅 Biopython wiki 上的 Phylo 页面 (https://biopython.pythonlang.cn/wiki/Phylo)。
PhyloXML 树的功能
phyloXML 文件格式包括用于用附加数据类型和视觉提示注释树的字段。
有关 PhyloXML 提供的附加注释功能的描述和示例,请参阅 Biopython wiki 上的 PhyloXML 页面 (https://biopython.pythonlang.cn/wiki/PhyloXML)。
运行外部应用程序
虽然 Bio.Phylo 本身不从比对中推断树,但有一些第三方程序可以做到这一点。可以使用 subprocess 模块从 python 中访问这些程序。
以下是如何使用 python 脚本与 PhyML (http://www.atgc-montpellier.fr/phyml/) 交互的示例。该程序接受 phylip-relaxed 格式的输入比对(即 Phylip 格式,但对分类单元名称没有 10 个字符的限制)以及各种选项。
>>> import subprocess
>>> cmd = "phyml -i Tests/Phylip/random.phy"
>>> results = subprocess.run(cmd, shell=True, stdout=subprocess.PIPE, text=True)
“stdout = subprocess.PIPE”参数使程序的输出可以通过“results.stdout”访问,用于调试目的(对“stderr”也可以执行相同的操作),而“text=True”使返回的信息成为 python 字符串,而不是“bytes”对象。
这将生成一个树文件和一个统计文件,其名称分别为 [输入文件名]_phyml_tree.txt 和 [输入文件名]_phyml_stats.txt。树文件采用 Newick 格式。
>>> from Bio import Phylo
>>> tree = Phylo.read("Tests/Phylip/random.phy_phyml_tree.txt", "newick")
>>> Phylo.draw_ascii(tree)
__________________ F
|
| I
|
_| ________ C
| ________|
| | | , J
| | |________|
| | | , H
|___________| |__________|
| |______________ D
|
, G
|
| , E
|________________|
| ___________________________ A
|________________|
|_________ B
subprocess 模块也可以用于与任何其他提供命令行界面的程序交互,例如 RAxML (https://sco.h-its.org/exelixis/software.html)、FastTree (http://www.microbesonline.org/fasttree/)、dnaml 和 protml。
PAML 集成
Biopython 1.58 带来了对 PAML (http://abacus.gene.ucl.ac.uk/software/paml.html) 的支持,PAML 是一个用于通过最大似然进行系统发育分析的程序套件。目前已实现 codeml、baseml 和 yn00 程序。由于 PAML 使用控制文件而不是命令行参数来控制运行时选项,因此此包装器的使用偏离了 Biopython 中其他应用程序包装器的格式。
典型的流程是初始化一个 PAML 对象,指定一个比对文件、一个树文件、一个输出文件和一个工作目录。接下来,通过 set_options() 方法设置运行时选项,或通过读取现有的控制文件来设置运行时选项。最后,通过 run() 方法运行程序,并将输出文件自动解析为结果字典。
以下是如何使用 codeml 的典型示例。
>>> from Bio.Phylo.PAML import codeml
>>> cml = codeml.Codeml()
>>> cml.alignment = "Tests/PAML/Alignments/alignment.phylip"
>>> cml.tree = "Tests/PAML/Trees/species.tree"
>>> cml.out_file = "results.out"
>>> cml.working_dir = "./scratch"
>>> cml.set_options(
... seqtype=1,
... verbose=0,
... noisy=0,
... RateAncestor=0,
... model=0,
... NSsites=[0, 1, 2],
... CodonFreq=2,
... cleandata=1,
... fix_alpha=1,
... kappa=4.54006,
... )
>>> results = cml.run()
>>> ns_sites = results.get("NSsites")
>>> m0 = ns_sites.get(0)
>>> m0_params = m0.get("parameters")
>>> print(m0_params.get("omega"))
使用模块的 read() 函数也可以解析现有的输出文件。
>>> results = codeml.read("Tests/PAML/Results/codeml/codeml_NSsites_all.out")
>>> print(results.get("lnL max"))
有关此新模块的详细文档目前位于 Biopython wiki 上:https://biopython.pythonlang.cn/wiki/PAML
未来计划
Bio.Phylo 正在积极开发中。以下是一些我们可能会在未来版本中添加的功能。
- 新方法
通常情况下,用于操作 Tree 或 Clade 对象的有用函数会先出现在 Biopython wiki 上,以便普通用户可以在我们将其添加到 Bio.Phylo 之前对其进行测试并决定其是否有用。
- Bio.Nexus 移植
该模块的很大一部分是在 2009 年的 Google Summer of Code 中编写的,在 NESCent 的支持下,作为实现对 phyloXML 数据格式的支持的项目(参见 PhyloXML 树的功能)。对 Newick 和 Nexus 格式的支持是通过将现有 Bio.Nexus 模块的一部分移植到 Bio.Phylo 使用的新类来实现的。
目前,Bio.Nexus 包含一些尚未移植到 Bio.Phylo 类中的有用功能——尤其是计算共识树。如果您发现 Bio.Phylo 中缺少一些功能,请尝试查看 Bio.Nexus,看看它是否在那里。
我们欢迎任何关于改进此模块的功能和可用性的建议;只需在邮件列表或我们的错误数据库中告知我们即可。
最后,如果您需要 Phylo 模块中尚未包含的附加功能,请检查它是否在其他高质量的 Python 系统发育库中可用,例如 DendroPy (https://dendropy.org/) 或 PyCogent (http://pycogent.org/)。由于这些库也支持系统发育树的标准文件格式,因此您可以通过写入临时文件或 StringIO 对象轻松地在库之间传输数据。