进入3D世界:PDB模块
Bio.PDB 是 Biopython 的一个模块,专注于处理生物大分子晶体结构。除其他事项外,Bio.PDB 包含一个 PDBParser 类,它会生成一个 Structure 对象,该对象可用于以便捷的方式访问文件中的原子数据。它对解析 PDB 头文件中包含的信息的支持有限。PDB 文件格式不再被修改或扩展以支持新内容,PDBx/mmCIF 在 2014 年成为标准的 PDB 档案格式。所有世界蛋白质数据库 (wwPDB) 站点都使用大分子晶体学信息文件 (mmCIF) 数据字典来描述 PDB 条目的信息内容。mmCIF 使用灵活且可扩展的键值对格式来表示大分子结构数据,并且对可以在单个 PDB 条目中表示的原子、残基或链的数量没有限制(没有拆分条目!)。
读取和写入晶体结构文件
读取 mmCIF 文件
首先创建一个 MMCIFParser 对象
>>> from Bio.PDB.MMCIFParser import MMCIFParser
>>> parser = MMCIFParser()
然后使用此解析器从 mmCIF 文件创建结构对象
>>> structure = parser.get_structure("1fat", "1fat.cif")
为了更低级地访问 mmCIF 文件,可以使用 MMCIF2Dict 类创建一个 Python 字典,该字典将 mmCIF 文件中的所有 mmCIF 标签映射到它们的值。无论是否有多个值(例如,标签 _atom_site.Cartn_y,它保存所有原子的 \(y\) 坐标)或单个值(例如,初始提交日期),该标签都映射到一个值列表。该字典是从 mmCIF 文件创建的,如下所示
>>> from Bio.PDB.MMCIF2Dict import MMCIF2Dict
>>> mmcif_dict = MMCIF2Dict("1FAT.cif")
示例:从 mmCIF 文件获取溶剂含量
>>> sc = mmcif_dict["_exptl_crystal.density_percent_sol"]
示例:获取所有原子的 \(y\) 坐标列表
>>> y_list = mmcif_dict["_atom_site.Cartn_y"]
读取二进制 CIF 文件
创建一个 BinaryCIFParser 对象
>>> from Bio.PDB.binary_cif import BinaryCIFParser
>>> parser = BinaryCIFParser()
使用指向二进制 CIF 文件的路径调用 get_structure
>>> parser.get_structure("1GBT", "1gbt.bcif.gz")
<Structure id=1GBT>
读取 MMTF 格式的文件
可以使用直接的 MMTFParser 从文件读取结构
>>> from Bio.PDB.mmtf import MMTFParser
>>> structure = MMTFParser.get_structure("PDB/4CUP.mmtf")
也可以使用相同的类通过 PDB ID 获取结构
>>> structure = MMTFParser.get_structure_from_url("4CUP")
这会为您提供一个结构对象,就像从 PDB 或 mmCIF 文件读取一样。
还可以使用 Biopython 在内部使用的外部 MMTF 库访问底层数据
>>> from mmtf import fetch
>>> decoded_data = fetch("4CUP")
例如,可以访问仅 X 坐标。
>>> print(decoded_data.x_coord_list)
读取 PDB 文件
首先创建一个 PDBParser 对象
>>> from Bio.PDB.PDBParser import PDBParser
>>> parser = PDBParser(PERMISSIVE=1)
PERMISSIVE 标志表示 PDB 文件中的一些常见问题(请参阅 示例)将被忽略(但请注意,一些原子和/或残基将缺失)。如果该标志不存在,如果在解析操作期间检测到任何问题,则会生成 PDBConstructionException。
然后通过让 PDBParser 对象解析 PDB 文件(在本例中,PDB 文件名为 pdb1fat.ent,1fat 是用户为结构定义的名称)来生成 Structure 对象
>>> structure_id = "1fat"
>>> filename = "pdb1fat.ent"
>>> structure = parser.get_structure(structure_id, filename)
可以使用 get_header 和 get_trailer 方法从 PDBParser 对象中提取 PDB 文件的头部和尾部(简单的字符串列表)。但请注意,许多 PDB 文件包含头部信息不完整或有错误。许多错误在等效的 mmCIF 文件中已得到修复。因此,如果您对头部信息感兴趣,最好使用上面描述的 ``MMCIF2Dict`` 工具从 mmCIF 文件中提取信息,而不是解析 PDB 头部。
现在已经澄清了,让我们回到解析 PDB 头部。结构对象有一个名为 header 的属性,它是一个 Python 字典,将头部记录映射到它们的值。
示例
>>> resolution = structure.header["resolution"]
>>> keywords = structure.header["keywords"]
可用的键是 name、head、deposition_date、release_date、structure_method、resolution、structure_reference(映射到一个引用列表)、journal_reference、author、compound(映射到包含关于结晶化合物的各种信息的字典)、has_missing_residues、missing_residues 和 astral(映射到包含关于域的附加信息的字典,如果存在)。
has_missing_residues 映射到一个布尔值,如果找到至少一行非空的 REMARK 465 头部行,则为 True。在这种情况下,您应该假设在实验中使用的分子有一些残基,其 ATOM 坐标无法确定。 missing_residues 映射到一个包含关于缺失残基的信息的字典列表。如果 PDB 头部不遵循 PDB 规范中的模板,则缺失残基列表将为空或不完整。
也可以在不创建 Structure 对象的情况下直接从 PDB 文件创建该字典,即
>>> from Bio.PDB import parse_pdb_header
>>> with open(filename, "r") as handle:
... header_dict = parse_pdb_header(handle)
...
读取 PQR 文件
为了解析 PQR 文件,请以与 PDB 文件类似的方式进行
创建一个 PDBParser 对象,使用 is_pqr 标志
>>> from Bio.PDB.PDBParser import PDBParser
>>> pqr_parser = PDBParser(PERMISSIVE=1, is_pqr=True)
is_pqr 标志设置为 True 表示要解析的文件是 PQR 文件,解析器应该读取每个原子条目的原子电荷和半径字段。按照与 PQR 文件相同的步骤,然后生成 Structure 对象,并解析 PQR 文件。
>>> structure_id = "1fat"
>>> filename = "pdb1fat.ent"
>>> structure = parser.get_structure(structure_id, filename, is_pqr=True)
读取 PDBML(PDB XML)文件
创建一个 PDBMLParser 对象
>>> from Bio.PDB.PDBMLParser import PDBMLParser
>>> pdbml_parser = PDBMLParser()
使用包含 XML 格式的 PDB 结构的文件路径或文件对象调用 get_structure
>>> structure = pdbml_parser.get_structure("1GBT.xml")
编写 mmCIF 文件
MMCIFIO 类可用于将结构写入 mmCIF 文件格式。
>>> io = MMCIFIO()
>>> io.set_structure(s)
>>> io.save("out.cif")
Select 类可以以类似于下面 PDBIO 的方式使用。使用 MMCIF2Dict 读取的 mmCIF 字典也可以写入。
>>> io = MMCIFIO()
>>> io.set_dict(d)
>>> io.save("out.cif")
编写 PDB 文件
为此,请使用 PDBIO 类。当然,也很容易写出结构的特定部分。
示例:保存结构
>>> io = PDBIO()
>>> io.set_structure(s)
>>> io.save("out.pdb")
如果要写出结构的一部分,请使用 Select 类(也在 PDBIO 中)。Select 有四种方法
accept_model(model)accept_chain(chain)accept_residue(residue)accept_atom(atom)
默认情况下,每种方法都返回 1(这意味着模型/链/残基/原子包含在输出中)。通过子类化 Select 并在适当的时候返回 0,您可以从输出中排除模型、链等。这可能很麻烦,但非常强大。以下代码只写出甘氨酸残基
>>> class GlySelect(Select):
... def accept_residue(self, residue):
... if residue.get_name() == "GLY":
... return True
... else:
... return False
...
>>> io = PDBIO()
>>> io.set_structure(s)
>>> io.save("gly_only.pdb", GlySelect())
如果您觉得这一切过于复杂,Dice 模块包含一个方便的 extract 函数,该函数写出链中从起始残基到结束残基的所有残基。
编写 PQR 文件
使用 PDBIO 类,就像您使用 PDB 文件一样,带有标志 is_pqr=True。PDBIO 方法也可以用于 PQR 文件。
示例:编写 PQR 文件
>>> io = PDBIO(is_pqr=True)
>>> io.set_structure(s)
>>> io.save("out.pdb")
编写 MMTF 文件
要将结构写入 MMTF 文件格式
>>> from Bio.PDB.mmtf import MMTFIO
>>> io = MMTFIO()
>>> io.set_structure(s)
>>> io.save("out.mmtf")
Select 类可以像上面一样使用。请注意,标准 MMTF 文件中包含的键合信息、二级结构分配和其他一些信息不会被写入,因为它们不容易从结构对象中确定。此外,在标准 MMTF 文件中分组到同一实体的分子被 MMTFIO 视为独立的实体。
结构表示
Structure 对象的整体布局遵循所谓的 SMCRA(结构/模型/链/残基/原子)架构。
一个结构包含模型。
一个模型包含链。
一条链包含残基。
一个残基包含原子。
这是许多结构生物学家/生物信息学家对结构的思考方式,它提供了一种简单但高效的方式来处理结构。当需要时,本质上会添加其他内容。Structure 对象的 UML 图(现在先忽略 Disordered 类)如 图 3 所示。这种数据结构不一定最适合表示结构的生物大分子内容,但对于很好地解释描述结构的文件(通常是 PDB 或 MMCIF 文件)中存在的数据绝对必要。如果此层次结构不能代表结构文件的内容,则可以肯定地说该文件包含错误,或者至少没有明确地描述结构。如果不能生成 SMCRA 数据结构,则有理由怀疑存在问题。因此,解析 PDB 文件可用于检测可能存在的问题。我们将在 示例 部分给出几个这方面的示例。
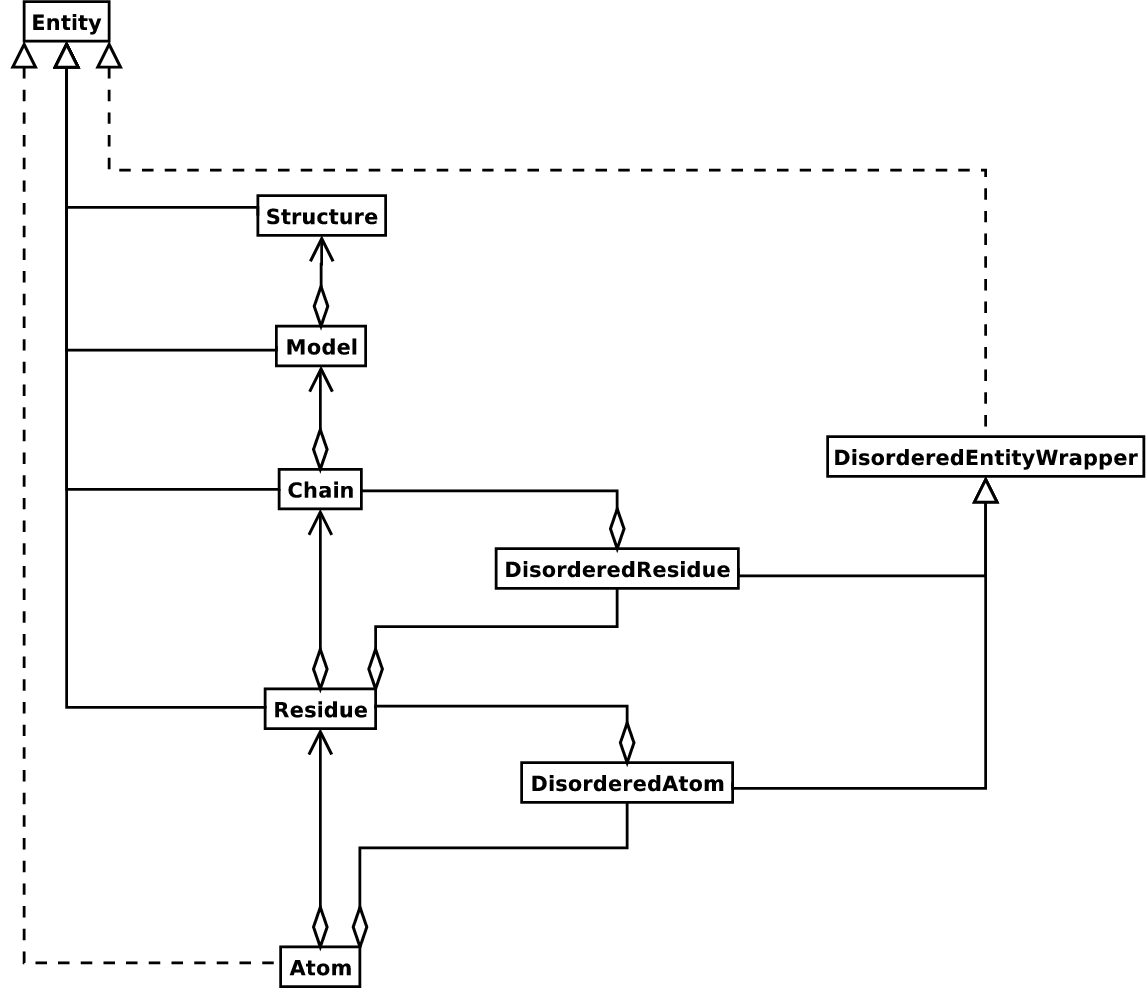
图 3 Structure 类的 SMCRA 架构的 UML 图。
这用于表示生物大分子结构。带菱形的实线表示聚合,带箭头的实线表示引用,带三角形的实线表示继承,带三角形的虚线表示接口实现。
Structure、Model、Chain 和 Residue 都是 Entity 基类的子类。Atom 类只(部分)实现了 Entity 接口(因为 Atom 没有子类)。
对于每个 Entity 子类,您可以使用该子类的唯一 ID 作为键来提取子类(例如,您可以使用原子名称字符串作为键从 Residue 对象中提取 Atom 对象,可以使用链标识符作为键从 Model 对象中提取 Chain 对象)。
无序原子和残基由 DisorderedAtom 和 DisorderedResidue 类表示,它们都是 DisorderedEntityWrapper 基类的子类。它们隐藏了与无序相关的复杂性,并且与 Atom 和 Residue 对象的行为完全相同。
一般来说,子 Entity 对象(即 Atom、Residue、Chain、Model)可以通过使用 ID 作为键从其父对象(分别为 Residue、Chain、Model、Structure)中提取。
>>> child_entity = parent_entity[child_id]
您还可以获取父 Entity 对象的所有子 Entity 对象的列表。请注意,此列表以特定方式排序(例如,根据 Model 对象中 Chain 对象的链标识符排序)。
>>> child_list = parent_entity.get_list()
您还可以从子对象中获取父对象。
>>> parent_entity = child_entity.get_parent()
在 SMCRA 层次结构的所有级别,您还可以提取完整 ID。完整 ID 是一个元组,包含从顶部对象(Structure)到当前对象的所有 ID。例如,Residue 对象的完整 ID 是
>>> full_id = residue.get_full_id()
>>> print(full_id)
("1abc", 0, "A", ("", 10, "A"))
这对应于
ID 为
"1abc"的 StructureID 为
0的 ModelID 为
"A"的 ChainID 为
("", 10, "A")的 Residue
Residue ID 表明该残基不是杂原子残基(也不是水),因为它有一个空白的杂原子字段,它的序列标识符是 10,并且它的插入代码是 "A"。
要获取实体的 ID,请使用 get_id 方法
>>> entity.get_id()
您可以使用 has_id 方法检查实体是否具有具有给定 ID 的子类
>>> entity.has_id(entity_id)
实体的长度等于其子类的数量。
>>> nr_children = len(entity)
可以从父实体中删除、重命名、添加等子实体,但这不包括任何健全性检查(例如,可以将两个具有相同 ID 的残基添加到同一个链中)。这确实应该通过一个包含完整性检查的漂亮装饰器类来完成,但如果您想使用原始接口,可以查看代码(Entity.py)。
Structure
Structure 对象位于层次结构的顶层。它的 ID 是用户给定的字符串。Structure 包含许多 Model 子类。大多数晶体结构(但并非全部)包含单个模型,而 NMR 结构通常包含多个模型。大型分子部分的晶体结构中的无序也可能导致多个模型。
Model
Model 对象的 ID 是一个整数,它源自模型在解析文件中出现的位置(它们从 0 开始自动编号)。晶体结构通常只有一个模型(ID 为 0),而 NMR 文件通常有多个模型。尽管许多 PDB 解析器假设只有一个模型,但 Bio.PDB 中的 Structure 类被设计成可以轻松处理包含多个模型的 PDB 文件。
例如,要从 Structure 对象中获取第一个模型,请使用
>>> first_model = structure[0]
Model 对象存储一个 Chain 子类列表。
Chain
Chain 对象的 ID 源自 PDB/mmCIF 文件中的链标识符,并且是一个单字符(通常是一个字母)。Model 对象中的每个 Chain 都具有唯一的 ID。例如,要从 Model 对象中获取标识符为“A”的 Chain 对象,请使用
>>> chain_A = model["A"]
Chain 对象存储一个 Residue 子类列表。
Residue
残基 ID 是一个包含三个元素的元组
杂原子字段 (hetfield):这是
'W',用于水分子;'H_'后跟其他杂原子残基的残基名称(例如,葡萄糖分子的'H_GLC');对于标准氨基酸和核酸,为空。
此方案是出于 相关问题 部分中描述的原因而采用的。
序列标识符 (resseq),一个整数,描述残基在链中的位置(例如,100);
插入代码 (icode);一个字符串,例如 'A'。插入代码有时用于保留某些理想的残基编号方案。例如,Ser 80 插入突变体(插入在 Thr 80 和 Asn 81 残基之间)可以具有以下序列标识符和插入代码:Thr 80 A、Ser 80 B、Asn 81。这样,残基编号方案与野生型结构的编号方案保持一致。
因此,上面葡萄糖残基的 ID 将是 (’H_GLC’, 100, ’A’)。如果杂原子标志和插入代码为空,则可以使用序列标识符本身
# Full id
>>> residue = chain[(" ", 100, " ")]
# Shortcut id
>>> residue = chain[100]
使用杂原子标志的原因是,许多 PDB 文件对氨基酸和杂原子残基或水使用相同的序列标识符,如果未使用杂原子标志,这会导致明显的问题。
毫无疑问,Residue 对象存储一组 Atom 子类。它还包含一个字符串,指定残基名称(例如,“ASN”)和残基的片段标识符(X-PLOR 用户熟知,但在构建 SMCRA 数据结构时未使用)。
让我们看一些例子。带有空白插入代码的 Asn 10 将具有残基 ID (’ ’, 10, ’ ’)。Water 10 将具有残基 ID (’W’, 10, ’ ’)。带有序列标识符 10 的葡萄糖分子(残基名为 GLC 的杂原子残基)将具有残基 ID (’H_GLC’, 10, ’ ’)。这样,三个残基(具有相同的插入代码和序列标识符)可以是同一条链的一部分,因为它们的残基 ID 不同。
在大多数情况下,hetflag 和插入代码字段将为空白,例如 (’ ’, 10, ’ ’)。 在这些情况下,序列标识符可以用作完整 id 的简写
# use full id
>>> res10 = chain[(" ", 10, " ")]
# use shortcut
>>> res10 = chain[10]
Chain 对象中的每个 Residue 对象都应该有一个唯一的 id。 然而,无序残基的处理方式有所不同,如第 点突变 部分所述。
Residue 对象有一些额外的属性
>>> residue.get_resname() # returns the residue name, e.g. "ASN"
>>> residue.is_disordered() # returns 1 if the residue has disordered atoms
>>> residue.get_segid() # returns the SEGID, e.g. "CHN1"
>>> residue.has_id(name) # test if a residue has a certain atom
您可以使用 is_aa(residue) 来测试 Residue 对象是否为氨基酸。
Atom
Atom 对象存储与原子相关联的数据,并且没有子级。 原子的 id 是它的原子名称(例如,Ser 残基的侧链氧的“OG”)。 Atom id 在 Residue 中需要是唯一的。 同样,对于无序原子,会做出例外,如第 无序原子 部分所述。
原子 id 只是原子名称(例如 ’CA’)。 实际上,原子名称是通过从 PDB 文件中的原子名称中去除所有空格创建的。
但是,在 PDB 文件中,空格可以是原子名称的一部分。 通常,钙原子被称为 ’CA..’ 以便将其与 C\(\alpha\) 原子(称为 ’.CA.’)区分开来。 在去除空格会导致问题(即同一个残基中存在两个名为 ’CA’ 的原子)的情况下,空格将保留。
在 PDB 文件中,原子名称由 4 个字符组成,通常带有前导和尾随空格。 通常,为了便于使用,可以删除这些空格(例如,氨基酸 C\(\alpha\) 原子在 PDB 文件中标记为“.CA.”,其中点代表空格)。 为了生成原子名称(以及原子 id),将删除空格,除非这会导致 Residue 中的名称冲突(即两个 Atom 对象具有相同的原子名称和 id)。 在后一种情况下,将尝试包括空格的原子名称。 例如,当一个残基包含名为“.CA.” 和“CA..” 的原子时,这种情况可能会发生,尽管这种情况不太可能。
存储的原子数据包括原子名称、原子坐标(如果存在则包括标准偏差)、B 因子(如果存在则包括各向异性 B 因子及其标准偏差)、altloc 说明符以及包含空格的完整原子名称。 不常用的项目,如 PDB 文件中有时指定的原子元素编号或原子电荷,不会被存储。
要操作原子坐标,请使用 Atom 对象的 transform 方法。 使用 set_coord 方法直接指定原子坐标。
Atom 对象具有以下附加方法
>>> a.get_name() # atom name (spaces stripped, e.g. "CA")
>>> a.get_id() # id (equals atom name)
>>> a.get_coord() # atomic coordinates
>>> a.get_vector() # atomic coordinates as Vector object
>>> a.get_bfactor() # isotropic B factor
>>> a.get_occupancy() # occupancy
>>> a.get_altloc() # alternative location specifier
>>> a.get_sigatm() # standard deviation of atomic parameters
>>> a.get_siguij() # standard deviation of anisotropic B factor
>>> a.get_anisou() # anisotropic B factor
>>> a.get_fullname() # atom name (with spaces, e.g. ".CA.")
要表示原子坐标,sigij、各向异性 B 因子和 sigatm Numpy 数组被使用。
The get_vector method 返回 Atom 对象坐标的 Vector 对象表示,允许您对原子坐标进行向量运算。 Vector 实现 3D 向量的完整集合,矩阵乘法(左乘和右乘)以及一些高级旋转相关运算。
作为 Bio.PDB 的 Vector 模块功能的示例,假设您想找到 Gly 残基的 C\(\beta\) 原子的位置,如果它存在的话。 将 Gly 残基的 N 原子沿 C\(\alpha\)-C 键旋转 -120 度,大致将其放置在虚拟 C\(\beta\) 原子的位置。 以下是操作方法,利用 Vector 模块的 rotaxis 方法(可用于构造绕特定轴的旋转)
# get atom coordinates as vectors
>>> n = residue["N"].get_vector()
>>> c = residue["C"].get_vector()
>>> ca = residue["CA"].get_vector()
# center at origin
>>> n = n - ca
>>> c = c - ca
# find rotation matrix that rotates n
# -120 degrees along the ca-c vector
>>> rot = rotaxis(-pi * 120.0 / 180.0, c)
# apply rotation to ca-n vector
>>> cb_at_origin = n.left_multiply(rot)
# put on top of ca atom
>>> cb = cb_at_origin + ca
此示例表明,可以在原子数据上进行一些相当非平凡的向量运算,这非常有用。 除了所有常见的向量运算(叉积(使用 **)和点积(使用 *)运算、角度、范数等)以及上面提到的 rotaxis 函数之外,Vector 模块还具有旋转 (rotmat) 或反射 (refmat) 一个向量到另一个向量上的方法。
从 Structure 中提取特定 Atom/Residue/Chain/Model
以下是一些示例
>>> model = structure[0]
>>> chain = model["A"]
>>> residue = chain[100]
>>> atom = residue["CA"]
请注意,您可以使用快捷方式
>>> atom = structure[0]["A"][100]["CA"]
Disorder
Bio.PDB 可以处理无序原子和点突变(即同一个位置的 Gly 和 Ala 残基)。
通用方法
无序应该从两个角度处理:原子和残基的角度。 一般来说,我们试图封装所有因无序而产生的复杂性。 如果您只想遍历所有 C\(\alpha\) 原子,则不关心某些残基是否有无序侧链。 另一方面,也应该能够在数据结构中完全表示无序。 因此,无序原子或残基存储在特殊对象中,这些对象的行为就像没有无序一样。 这是通过仅表示无序原子或残基的子集来完成的。 用户可以指定选择哪个子集(例如,使用 Ser 残基的两个无序 OG 侧链原子位置中的哪个)。
无序原子
无序原子由普通的 Atom 对象表示,但是所有表示相同物理原子的 Atom 对象都存储在 DisorderedAtom 对象中(参见 图 3)。 DisorderedAtom 对象中的每个 Atom 对象可以使用其 altloc 说明符进行唯一索引。 DisorderedAtom 对象将所有未捕获的属性调用转发到选定的 Atom 对象,默认情况下是表示占用率最高的原子的那个。 当然,用户可以更改选定的 Atom 对象,利用其 altloc 说明符。 以这种方式,原子无序以不增加太多复杂性的方式正确表示。 换句话说,如果您对原子无序不感兴趣,您将不会受到它的困扰。
每个无序原子都有一个特征性的 altloc 标识符。 您可以指定 DisorderedAtom 对象应该表现得像与特定 altloc 标识符相关联的 Atom 对象
>>> atom.disordered_select("A") # select altloc A atom
>>> print(atom.get_altloc())
"A"
>>> atom.disordered_select("B") # select altloc B atom
>>> print(atom.get_altloc())
"B"
无序残基
常见情况
最常见的情况是包含一个或多个无序原子的残基。 这显然是通过使用 DisorderedAtom 对象来表示无序原子,并将 DisorderedAtom 对象存储在 Residue 对象中(就像普通的 Atom 对象一样)来解决的。 DisorderedAtom 的行为将与普通原子完全相同(实际上是占用率最高的原子),通过将所有未捕获的属性调用转发到它包含的 Atom 对象之一(选定的 Atom 对象)。
点突变
当无序是由于点突变造成的时,会出现一个特殊情况,即当多肽的两个或多个点突变体存在于晶体中时。 在 PDB 结构 1EN2 中可以找到这种情况的示例。
由于这些残基属于不同的残基类型(例如,假设是 Ser 60 和 Cys 60),因此它们不应该像在常见情况下那样存储在单个 Residue 对象中。 在这种情况下,每个残基由一个 Residue 对象表示,并且两个 Residue 对象存储在单个 DisorderedResidue 对象中(参见 图 3)。
The DisorderedResidue 对象将所有未捕获的属性调用转发到选定的 Residue 对象(默认情况下是最后一个添加的 Residue 对象),因此其行为就像普通的残基一样。 DisorderedResidue 对象中的每个 Residue 对象可以使用其残基名称进行唯一标识。 在上面的示例中,残基 Ser 60 在 DisorderedResidue 对象中的 id 为“SER”,而残基 Cys 60 的 id 为“CYS”。 用户可以通过此 id 选择 DisorderedResidue 对象中的活动 Residue 对象。
示例:假设一条链在位置 10 处有一个点突变,由 Ser 和 Cys 残基组成。 确保该链的残基 10 的行为与 Cys 残基相同。
>>> residue = chain[10]
>>> residue.disordered_select("CYS")
此外,您可以使用 (Disordered)Residue 对象的 get_unpacked_list 方法获取所有 Atom 对象的列表(即所有 DisorderedAtom 对象都“解包”到它们各自的 Atom 对象中)。
杂原子
相关问题
杂原子常见问题是,同一链中的多个杂原子和非杂原子残基共享相同的序列标识符(和插入代码)。 因此,为了为每个杂原子生成一个唯一的 id,水和其他杂原子的处理方式不同。
请记住,Residue 对象的 id 是元组 (hetfield, resseq, icode)。 对于氨基酸和核酸,hetfield 为空白 (“ ”),对于水和其他杂原子,hetfield 为字符串。 hetfield 的内容将在下面解释。
水残基
水残基的 hetfield 字符串由字母“W”组成。 因此,水的典型残基 id 为 (“W”, 1, “ ”)。
其他杂原子
其他杂原子的 hetfield 字符串以“H_”开头,后跟残基名称。 例如,残基名称为“GLC”的葡萄糖分子将具有 hetfield“H_GLC”。 其残基 id 可能例如为 (“H_GLC”, 1, “ ”)。
分析结构
测量距离
原子的减法运算符已被重载以返回两个原子之间的距离。
# Get some atoms
>>> ca1 = residue1["CA"]
>>> ca2 = residue2["CA"]
# Simply subtract the atoms to get their distance
>>> distance = ca1 - ca2
测量角度
使用原子坐标的向量表示,以及 Vector 模块中的 calc_angle 函数
>>> vector1 = atom1.get_vector()
>>> vector2 = atom2.get_vector()
>>> vector3 = atom3.get_vector()
>>> angle = calc_angle(vector1, vector2, vector3)
测量扭转角
使用原子坐标的向量表示,以及 Vector 模块中的 calc_dihedral 函数
>>> vector1 = atom1.get_vector()
>>> vector2 = atom2.get_vector()
>>> vector3 = atom3.get_vector()
>>> vector4 = atom4.get_vector()
>>> angle = calc_dihedral(vector1, vector2, vector3, vector4)
内部坐标 - 距离、角度、扭转角、距离图等
蛋白质结构通常以相对于固定原点的 3D XYZ 坐标形式提供,如 PDB 或 mmCIF 文件中。 internal_coords 模块有助于将此系统在键长、角度和二面角之间进行转换。除了支持对蛋白质结构进行标准的 _psi、phi、chi_ 等计算外,此表示对平移和旋转是不变的,并且该实现展现了结构分析的多种优势。
首先在这里加载一些模块以备后续示例使用
>>> from Bio.PDB.PDBParser import PDBParser
>>> from Bio.PDB.Chain import Chain
>>> from Bio.PDB.internal_coords import *
>>> from Bio.PDB.PICIO import write_PIC, read_PIC, read_PIC_seq
>>> from Bio.PDB.ic_rebuild import write_PDB, IC_duplicate, structure_rebuild_test
>>> from Bio.PDB.SCADIO import write_SCAD
>>> from Bio.Seq import Seq
>>> from Bio.SeqRecord import SeqRecord
>>> from Bio.PDB.PDBIO import PDBIO
>>> import numpy as np
访问二面角、角度和键长
我们从计算结构的内部坐标的简单情况开始
>>> # load a structure as normal, get first chain
>>> parser = PDBParser()
>>> myProtein = parser.get_structure("1a8o", "1A8O.pdb")
>>> myChain = myProtein[0]["A"]
>>> # compute bond lengths, angles, dihedral angles
>>> myChain.atom_to_internal_coordinates(verbose=True)
chain break at THR 186 due to MaxPeptideBond (1.4 angstroms) exceeded
chain break at THR 216 due to MaxPeptideBond (1.4 angstroms) exceeded
通过删除上面的 verbose=True 选项,1A8O 的链断裂警告被抑制。要避免创建断裂,而是允许不切实际的 N-C 键长,请覆盖类变量 MaxPeptideBond,例如
>>> IC_Chain.MaxPeptideBond = 4.0
>>> myChain.internal_coord = None # force re-loading structure data with new cutoff
>>> myChain.atom_to_internal_coordinates(verbose=True)
此时,这些值可以在链级和残基级使用。1A8O 的第一个残基是 HETATM MSE(硒代蛋氨酸),因此我们使用规范名称或原子说明符检查下面的残基 2。我们通过名称和原子序列获得 _chi1_ 二面角和 _tau_ 角,并通过指定原子对获得 C\(\alpha\)-C\(\beta\) 距离
>>> r2 = myChain.child_list[1]
>>> r2
<Residue ASP het= resseq=152 icode= >
>>> r2ic = r2.internal_coord
>>> print(r2ic, ":", r2ic.pretty_str(), ":", r2ic.rbase, ":", r2ic.lc)
('1a8o', 0, 'A', (' ', 152, ' ')) : ASP 152 : (152, None, 'D') : D
>>> r2chi1 = r2ic.get_angle("chi1")
>>> print(round(r2chi1, 2))
-144.86
>>> r2ic.get_angle("chi1") == r2ic.get_angle("N:CA:CB:CG")
True
>>> print(round(r2ic.get_angle("tau"), 2))
113.45
>>> r2ic.get_angle("tau") == r2ic.get_angle("N:CA:C")
True
>>> print(round(r2ic.get_length("CA:CB"), 2))
1.53
Chain.internal_coord 对象包含 hedra(3 个键合原子)和 dihedra(4 个键合原子)对象的数组和字典。字典按 AtomKey 对象元组索引;AtomKey 对象捕获残基位置、插入代码、1 或 3 个字符的残基名称、原子名称、altloc 和占据率。
下面,我们通过直接索引 Chain 数组获得与上面相同的 _chi1_ 和 _tau_ 角,使用 AtomKey 来索引 Chain 数组
>>> myCic = myChain.internal_coord
>>> r2chi1_object = r2ic.pick_angle("chi1")
>>> # or same thing (as for get_angle() above):
>>> r2chi1_object == r2ic.pick_angle("N:CA:CB:CG")
True
>>> r2chi1_key = r2chi1_object.atomkeys
>>> r2chi1_key # r2chi1_key is tuple of AtomKeys
(152_D_N, 152_D_CA, 152_D_CB, 152_D_CG)
>>> r2chi1_index = myCic.dihedraNdx[r2chi1_key]
>>> # or same thing:
>>> r2chi1_index == r2chi1_object.ndx
True
>>> print(round(myCic.dihedraAngle[r2chi1_index], 2))
-144.86
>>> # also:
>>> r2chi1_object == myCic.dihedra[r2chi1_key]
True
>>> # hedra angles are similar:
>>> r2tau = r2ic.pick_angle("tau")
>>> print(round(myCic.hedraAngle[r2tau.ndx], 2))
113.45
在 Chain 级获取键长数据更为复杂(不推荐)。如这里所示,多个 hedra 将在不同的位置共享一个键
>>> r2CaCb = r2ic.pick_length("CA:CB") # returns list of hedra containing bond
>>> r2CaCb[0][0].atomkeys
(152_D_CB, 152_D_CA, 152_D_C)
>>> print(round(myCic.hedraL12[r2CaCb[0][0].ndx], 2)) # position 1-2
1.53
>>> r2CaCb[0][1].atomkeys
(152_D_N, 152_D_CA, 152_D_CB)
>>> print(round(myCic.hedraL23[r2CaCb[0][1].ndx], 2)) # position 2-3
1.53
>>> r2CaCb[0][2].atomkeys
(152_D_CA, 152_D_CB, 152_D_CG)
>>> print(round(myCic.hedraL12[r2CaCb[0][2].ndx], 2)) # position 1-2
1.53
请改用 Residue 级的 set_length:math:`` 函数。
测试结构的完整性
缺失原子和其他问题可能会在重建结构时导致问题。使用 structure_rebuild_test:math:`` 来快速确定结构是否具有足够的数据以进行干净重建。添加 verbose=True 和/或检查结果字典以获取更多详细信息
>>> # check myChain makes sense (can get angles and rebuild same structure)
>>> resultDict = structure_rebuild_test(myChain)
>>> resultDict["pass"]
True
修改和重建结构
最好使用残基级的 set_angle:math:`` 和 set_length:math:`` 功能来修改内部坐标,而不是直接访问 Chain 结构。虽然直接修改 hedra 角是安全的,但键长出现在多个重叠的 hedra 中,如上所述,这由 set_length:math:. 当 应用 于 二面角 时, ``set_angle:math:`` 会将结果包装到 +/-180 并旋转相邻的二面角(例如异亮氨酸 _chi1_ 角的两个键 - 这可能就是您想要的)。
>>> # rotate residue 2 chi1 angle by -120 degrees
>>> r2ic.set_angle("chi1", r2chi1 - 120.0)
>>> print(round(r2ic.get_angle("chi1"), 2))
95.14
>>> r2ic.set_length("CA:CB", 1.49)
>>> print(round(myCic.hedraL12[r2CaCb[0][0].ndx], 2)) # Cb-Ca-C position 1-2
1.49
从内部坐标重建结构是对 internal_to_atom_coordinates() 的简单调用
>>> myChain.internal_to_atom_coordinates()
>>> # just for proof:
>>> myChain.internal_coord = None # all internal_coord data removed, only atoms left
>>> myChain.atom_to_internal_coordinates() # re-generate internal coordinates
>>> r2ic = myChain.child_list[1].internal_coord
>>> print(round(r2ic.get_angle("chi1"), 2)) # show measured values match what was set above
95.14
>>> print(round(myCic.hedraL23[r2CaCb[0][1].ndx], 2)) # N-Ca-Cb position 2-3
1.49
生成的结构可以用 PDBIO 正常写入
write_PDB(myProtein, "myChain.pdb")
# or just the ATOM records without headers:
io = PDBIO()
io.set_structure(myProtein)
io.save("myChain2.pdb")
蛋白质内部坐标 (.pic) 文件和默认值
PICIO 模块中定义了一种文件格式,用于将蛋白质链描述为相对于初始坐标的 hedra 和 dihedra。除了残基序列信息之外的所有文件部分(例如 (’1A8O’, 0, ’A’, (’ ’, 153, ’ ’)) ILE)都是可选的,如果未指定并且 read_PIC:math:`` 使用 defaults=True 选项调用,则会使用默认值填充。默认值是从 2019 年 9 月的 Dunbrack cullpdb_pc20_res2.2_R1.0 计算得出的。
在这里,我们将 “myChain” 编写为 .pic 内部坐标规范文件,然后将其读回作为 “myProtein2”。
# write chain as 'protein internal coordinates' (.pic) file
write_PIC(myProtein, "myChain.pic")
# read .pic file
myProtein2 = read_PIC("myChain.pic")
由于所有内部坐标值都可以用默认值替换,因此提供了 PICIO.read_PIC_seq:math:`` 作为实用程序函数,以从输入序列创建有效的(主要是螺旋形的)默认结构
# create default structure for random sequence by reading as .pic file
myProtein3 = read_PIC_seq(
SeqRecord(
Seq("GAVLIMFPSTCNQYWDEHKR"),
id="1RND",
description="my random sequence",
)
)
myProtein3.internal_to_atom_coordinates()
write_PDB(myProtein3, "myRandom.pdb")
您可能希望探索在从内部坐标生成结构时,例如 _omega_ 角 (180.0)、hedra 角和/或键长的精度要求。 write_PIC:math:`` 的 picFlags 选项可以实现这一点,允许选择要写入 .pic 文件的数据,而未指定的数据将获得默认值。
各种组合都是可能的,并提供了一些预设,例如 classic 仅将 _psi、phi、tau_、脯氨酸 _omega_ 和侧链 _chi_ 角写入 .pic 文件
write_PIC(myProtein, "myChain.pic", picFlags=IC_Residue.pic_flags.classic)
myProtein2 = read_PIC("myChain.pic", defaults=True)
访问全原子 AtomArray
Biopython Atom 对象中的所有 3D XYZ 坐标在 Chain 类中移动到一个大型数组,并在 atom_to_internal_coordinates:math:. 访问 Biopython ``Atom 坐标的软件不受影响,但新数组可能为将来的工作提供效率。
与 Atom 的 XYZ 坐标不同,AtomArray 坐标是同构的,这意味着它们是类似于 [ x y z 1.0] 的数组,其中 1.0 是第四个元素。这方便了在整个 internal_coords 模块中使用组合的平移和旋转矩阵进行高效变换。还有一个相应的 AtomArrayIndex 字典,将 AtomKeys 映射到它们的坐标。
这里我们演示了从数组中读取特定 C\(\beta\) 原子的坐标,然后展示修改数组值会同时修改 Atom 对象。
>>> # access the array of all atoms for the chain, e.g. r2 above is residue 152 C-beta
>>> r2_cBeta_index = myChain.internal_coord.atomArrayIndex[AtomKey("152_D_CB")]
>>> r2_cBeta_coords = myChain.internal_coord.atomArray[r2_cBeta_index]
>>> print(np.round(r2_cBeta_coords, 2))
[-0.75 -1.18 -0.51 1. ]
>>> # the Biopython Atom coord array is now a view into atomArray, so
>>> assert r2_cBeta_coords[1] == r2["CB"].coord[1]
>>> r2_cBeta_coords[1] += 1.0 # change the Y coord 1 angstrom
>>> assert r2_cBeta_coords[1] == r2["CB"].coord[1]
>>> # they are always the same (they share the same memory)
>>> r2_cBeta_coords[1] -= 1.0 # restore
请注意,很容易“破坏”Atom 坐标数组和链 atomArray 之间的视图链接。当直接修改 Atom 坐标时,请使用逐元素复制的语法来避免这种情况。
# use these:
myAtom1.coord[:] = myAtom2.coord
myAtom1.coord[...] = myAtom2.coord
myAtom1.coord[:] = [1, 2, 3]
for i in range(3):
myAtom1.coord[i] = myAtom2.coord[i]
# do not use:
myAtom1.coord = myAtom2.coord
myAtom1.coord = [1, 2, 3]
使用 atomArrayIndex 和对 AtomKey 类的了解,我们可以创建 Numpy “选择器”,如下所示,以提取仅包含 C\(\alpha\) 原子坐标的数组。
>>> # create a selector to filter just the C-alpha atoms from the all atom array
>>> atmNameNdx = AtomKey.fields.atm
>>> aaI = myChain.internal_coord.atomArrayIndex
>>> CaSelect = [aaI.get(k) for k in aaI.keys() if k.akl[atmNameNdx] == "CA"]
>>> # now the ordered array of C-alpha atom coordinates is:
>>> CA_coords = myChain.internal_coord.atomArray[CaSelect]
>>> # note this uses Numpy fancy indexing, so CA_coords is a new copy
>>> # (if you modify it, the original atomArray is unaffected)
距离图
atomArray 的一个优势是,从它生成距离图只需一行 Numpy 代码。
np.linalg.norm(atomArray[:, None, :] - atomArray[None, :, :], axis=-1)
尽管很简短,但这种习惯用法可能难以记忆,并且在上面的形式中生成了所有原子的距离,而不是经典的 C\(\alpha\) 图,这可能是人们想要的。 distance_plot:math:`` 方法包装了上面的代码行,并接受一个可选的选择器,例如上一节中定义的 CaSelect。参见 图 4.
# create a C-alpha distance plot
caDistances = myChain.internal_coord.distance_plot(CaSelect)
# display with e.g. MatPlotLib:
import matplotlib.pyplot as plt
plt.imshow(caDistances, cmap="hot", interpolation="nearest")
plt.show()
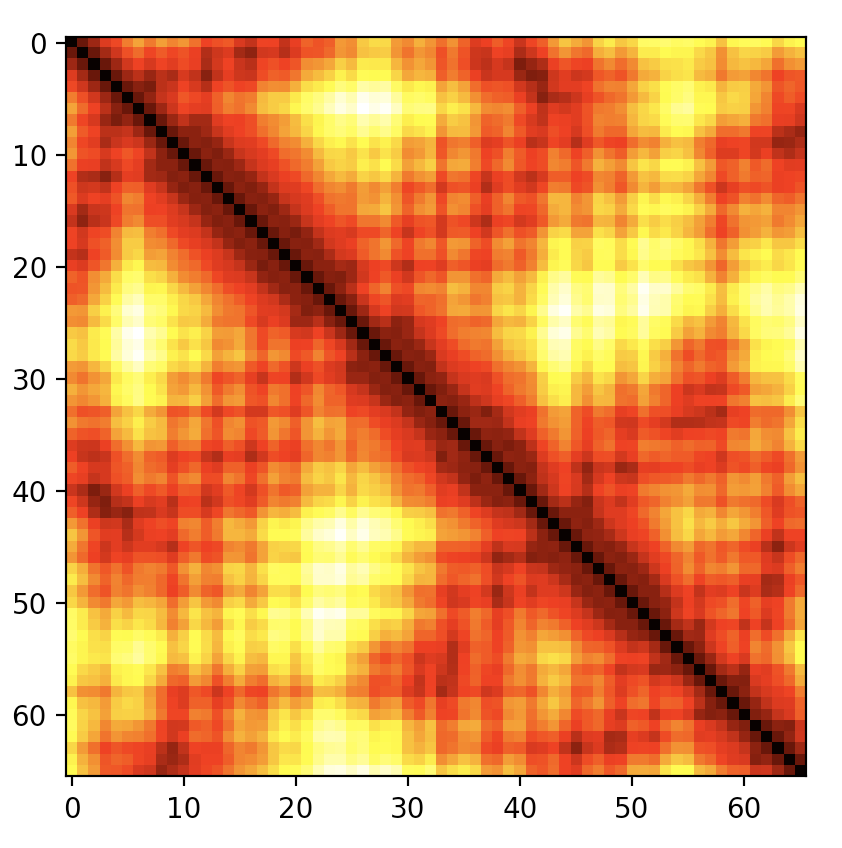
图 4 PDB 文件 1A8O (HIV 衣壳 C 端结构域) 的 C-alpha 距离图
从距离图构建结构
所有原子距离图是蛋白质结构的另一种表示,它也与平移和旋转无关,但缺乏手性信息(镜像结构将生成相同的距离图)。通过将距离矩阵与每个二面角的符号相结合,可以重新生成内部坐标。
这项工作使用 Blue 开发的方程,即 Hedronometer,在 https://math.stackexchange.com/a/49340/409 中讨论,并在 http://daylateanddollarshort.com/mathdocs/Heron-like-Results-for-Tetrahedral-Volume.pdf 中进一步讨论。
首先,我们从“myChain”中提取距离和手性值。
>>> ## create the all-atom distance plot
>>> distances = myCic.distance_plot()
>>> ## get the signs of the dihedral angles
>>> chirality = myCic.dihedral_signs()
我们需要一个与“myChain”匹配的有效数据结构来正确重建它;使用上面的 read_PIC_seq:math:`` 在一般情况下会起作用,但这里使用的 1A8O 示例具有一些 ALTLOC 复杂性,而序列本身无法生成。为了演示,最简单的方法是简单地复制“myChain”结构,但我们将所有原子和内部坐标链数组设置为 0(仅用于演示),以确保没有数据来自原始结构。
>>> ## get new, empty data structure : copy data structure from myChain
>>> myChain2 = IC_duplicate(myChain)[0]["A"]
>>> cic2 = myChain2.internal_coord
>>> ## clear the new atomArray and di/hedra value arrays, just for proof
>>> cic2.atomArray = np.zeros((cic2.AAsiz, 4), dtype=np.float64)
>>> cic2.dihedraAngle[:] = 0.0
>>> cic2.hedraAngle[:] = 0.0
>>> cic2.hedraL12[:] = 0.0
>>> cic2.hedraL23[:] = 0.0
该方法是从距离图数据重新生成内部坐标,然后从内部坐标生成原子坐标,如上所示。为了将最终生成的结构放置在与起始结构相同的坐标空间中,我们将“myChain”链开头的第一个三个 N-C\(\alpha\)-C 原子的坐标复制到“myChain2”结构中(这仅用于演示结束时的等价性)。
>>> ## copy just the first N-Ca-C coords so structures will superimpose:
>>> cic2.copy_initNCaCs(myChain.internal_coord)
distance_to_internal_coordinates:math:`` 例程需要目标结构中每个二面体六个原子间距离的数组。便捷例程 distplot_to_dh_arrays:math:`` 从先前生成的距离矩阵中提取这些值,可以由用户方法替换,以将这些数据写入 Chain.internal_coords 对象中的数组。
>>> ## copy distances to chain arrays:
>>> cic2.distplot_to_dh_arrays(distances, chirality)
>>> ## compute angles and dihedral angles from distances:
>>> cic2.distance_to_internal_coordinates()
以下步骤从新生成的“myChain2”内部坐标生成原子坐标,然后使用 Numpy allclose:math:`` 例程确认所有值与 PDB 文件分辨率的匹配度。
>>> ## generate XYZ coordinates from internal coordinates:
>>> myChain2.internal_to_atom_coordinates()
>>> ## confirm result atomArray matches original structure:
>>> np.allclose(cic2.atomArray, myCic.atomArray)
True
请注意,此过程不会使用整个距离矩阵,而只使用每个二面体四个原子之间的六个局部距离。
叠加残基及其邻域
internal_coords 模块依赖于在不同坐标空间之间转换原子坐标,用于计算扭转角和重建结构。每个二面体都有一个坐标空间变换,将它的第一个原子放在 XZ 平面上,第二个原子放在原点上,第三个原子放在 +Z 轴上,以及一个相应的反向变换,它将把它返回到原始结构中的坐标。这些变换矩阵可以像下面这样使用。通过明智地选择参考二面体,可以对多个蛋白质结构中的成对和更高阶的残基相互作用进行研究和可视化,例如 图 5.
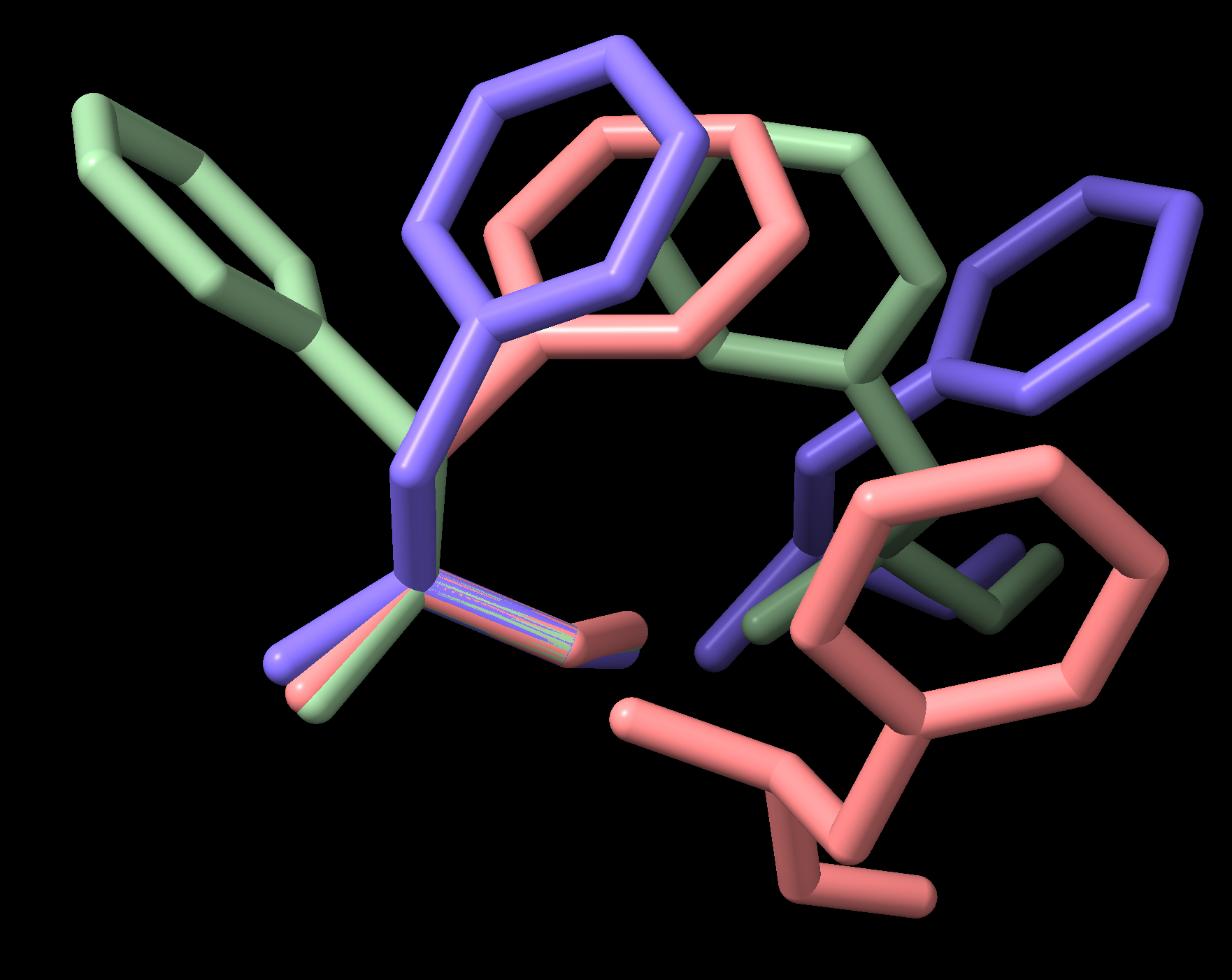
图 5 PDB 文件 3PBL (人类多巴胺 D3 受体) 中的相邻苯丙氨酸侧链
此示例将链中每个 PHE 残基在其 N-C\(\alpha\)-C\(\beta\) 原子上叠加,并在相应的坐标空间中呈现链中的所有 PHE,作为一个简单的演示。对成对侧链相互作用的更现实的探索将检查结构数据集并过滤与相关文献中讨论的相互作用类别。
# superimpose all phe-phe pairs - quick hack just to demonstrate concept
# for analyzing pairwise residue interactions. Generates PDB ATOM records
# placing each PHE at origin and showing all other PHEs in environment
## shorthand for key variables:
cic = myChain.internal_coord
resNameNdx = AtomKey.fields.resname
aaNdx = cic.atomArrayIndex
## select just PHE atoms:
pheAtomSelect = [aaNdx.get(k) for k in aaNdx.keys() if k.akl[resNameNdx] == "F"]
aaF = cic.atomArray[pheAtomSelect] # numpy fancy indexing makes COPY not view
for ric in cic.ordered_aa_ic_list: # internal_coords version of get_residues()
if ric.lc == "F": # if PHE, get transform matrices for chi1 dihedral
chi1 = ric.pick_angle("chi1") # N:CA:CB:CG space has C-alpha at origin
cst = np.transpose(chi1.cst) # transform TO chi1 space
# rcst = np.transpose(chi1.rcst) # transform FROM chi1 space (not needed here)
cic.atomArray[pheAtomSelect] = aaF.dot(cst) # transform just the PHEs
for res in myChain.get_residues(): # print PHEs in new coordinate space
if res.resname in ["PHE"]:
print(res.internal_coord.pdb_residue_string())
cic.atomArray[pheAtomSelect] = aaF # restore coordinate space from copy
3D 打印蛋白质结构
OpenSCAD (https://openscad.org) 是一种用于创建实体 3D CAD 对象的语言。从内部坐标构建蛋白质结构的算法在 OpenSCAD 中提供,并附带描述结构的数据,以便可以生成适合 3D 打印的模型。虽然其他软件可以将 STL 数据作为 3D 打印的渲染选项(例如 Chimera,https://www.cgl.ucsf.edu/chimera/),但这种方法将球体和圆柱体作为输出,因此更适合于与 3D 打印蛋白质结构相关的修改。可以在 OpenSCAD 代码中选择单个残基和键以进行特殊处理,例如通过大小突出显示或在特定位置添加可旋转键(参见 https://www.thingiverse.com/thing:3957471 了解示例)。
# write OpenSCAD program of spheres and cylinders to 3d print myChain backbone
## set atom load filter to accept backbone only:
IC_Residue.accept_atoms = IC_Residue.accept_backbone
## set chain break cutoff very high to bridge missing residues with long bonds
IC_Chain.MaxPeptideBond = 4.0
## delete existing data to force re-read of all atoms with attributes set above:
myChain.internal_coord = None
write_SCAD(myChain, "myChain.scad", scale=10.0)
internal_coords 控制属性
internal_coords 类中提供了一些控制属性来修改或过滤数据,因为内部坐标是计算出来的。这些列在表 Bio.PDB.internal_coords 中的控制属性.
类 |
属性 |
默认值 |
影响 |
|---|---|---|---|
AtomKey |
d2h |
False |
如果为 True,则将 D 原子转换为 H |
IC_Chain |
MaxPeptideBond |
1.4 |
没有链断裂的最大 C-N 长度;设置为较大的值可以连接缺失的残基以用于 3D 模型 |
IC_Residue |
accept_atoms |
主链,氢原子 |
覆盖以移除一些或所有侧链、H、D |
IC_Residue |
accept_resnames |
CYG、YCM、UNK |
要处理的 HETATM 的 3 个字母名称,仅处理主链,除非添加到 ic_data.py 中 |
IC_Residue |
gly_Cbeta |
False |
覆盖以根据数据库平均值生成 Gly C\(\beta\) 原子 |
确定原子-原子接触
使用 NeighborSearch 执行邻居查找。邻居查找使用用 C 编写的 KD 树模块完成(参见模块 Bio.PDB.kdtrees 中的 KDTree 类),使其非常快。它还包含一种快速方法,用于查找彼此之间距离在特定距离内的所有点对。
叠加两个结构
使用 Superimposer 对象叠加两个坐标集。此对象计算旋转和平移矩阵,它以最小化其 RMSD 的方式将两个原子列表旋转到彼此之上。当然,这两个列表需要包含相同数量的原子。 Superimposer 对象也可以将旋转/平移应用于原子列表。旋转和平移存储为 Superimposer 对象的 rotran 属性中的元组(请注意,旋转是右乘的!)。RMSD 存储在 rmsd 属性中。
Superimposer 使用的算法来自 Golub & Van Loan [Golub1989],并利用奇异值分解(这在通用 Bio.SVDSuperimposer 模块中实现)。
示例
>>> sup = Superimposer()
# Specify the atom lists
# 'fixed' and 'moving' are lists of Atom objects
# The moving atoms will be put on the fixed atoms
>>> sup.set_atoms(fixed, moving)
# Print rotation/translation/rmsd
>>> print(sup.rotran)
>>> print(sup.rms)
# Apply rotation/translation to the moving atoms
>>> sup.apply(moving)
要根据活性位点叠加两个结构,请使用活性位点原子计算旋转/平移矩阵(如上所示),并将这些矩阵应用于整个分子。
计算半球暴露
半球暴露 (HSE) 是一种新的 2D 溶剂暴露度量 [Hamelryck2005]。基本上,它计算残基周围的 C\(\alpha\) 原子数量,这些原子在侧链方向和相反方向(在 13 Å 的半径内)。尽管很简单,但它优于许多其他溶剂暴露度量。
HSE 有两种形式:HSE\(\alpha\) 和 HSE\(\beta\)。前者只使用 C\(\alpha\) 原子位置,而后者使用 C\(\alpha\) 和 C\(\beta\) 原子位置。HSE 度量由 HSExposure 类计算,该类还可以计算接触数。后者类的方法返回字典,将 Residue 对象映射到其相应的 HSE\(\alpha\)、HSE\(\beta\) 和接触数值。
示例
>>> model = structure[0]
>>> hse = HSExposure()
# Calculate HSEalpha
>>> exp_ca = hse.calc_hs_exposure(model, option="CA3")
# Calculate HSEbeta
>>> exp_cb = hse.calc_hs_exposure(model, option="CB")
# Calculate classical coordination number
>>> exp_fs = hse.calc_fs_exposure(model)
# Print HSEalpha for a residue
>>> print(exp_ca[some_residue])
确定二级结构
要使用此功能,您需要安装 DSSP(并获取许可证——学术用途免费,请参阅 https://swift.cmbi.umcn.nl/gv/dssp/)。然后使用 DSSP 类,它将 Residue 对象映射到它们的二级结构(以及可及表面积)。DSSP 代码列于表 Bio.PDB 中的 DSSP 代码。 中。请注意,DSSP(程序,以及因此的结果类)无法处理多个模型!
代码 |
二级结构 |
|---|---|
H |
\(\alpha\)-螺旋 |
B |
孤立的 \(\beta\)-桥残基 |
E |
链 |
G |
3-10 螺旋 |
I |
\(\Pi\)-螺旋 |
T |
转角 |
S |
弯曲 |
其他 |
DSSP 类还可以用来计算残基的可及表面积。但也可以参考部分 计算残基深度。
计算残基深度
残基深度是指残基原子到溶剂可及表面的平均距离。这是一个相当新的,并且非常有效的溶剂可及性参数化方法。要使用此功能,您需要安装 Michel Sanner 的 MSMS 程序 (https://www.scripps.edu/sanner/html/msms_home.html)。然后使用 ResidueDepth 类。此类作为字典,它将 Residue 对象映射到相应的(残基深度,C\(\alpha\) 深度)元组。C\(\alpha\) 深度是指残基的 C\(\alpha\) 原子到溶剂可及表面的距离。
示例
>>> model = structure[0]
>>> rd = ResidueDepth(model, pdb_file)
>>> residue_depth, ca_depth = rd[some_residue]
您还可以访问分子表面本身(通过 get_surface 函数),它以包含表面点的 Numeric Python 数组的形式存在。
PDB 文件中的常见问题
众所周知,许多 PDB 文件包含语义错误(不是结构本身,而是它们在 PDB 文件中的表示)。Bio.PDB 尝试通过两种方式来处理这个问题。PDBParser 对象可以有两种行为方式:严格方式和宽松方式,后者是默认方式。
示例
# Permissive parser
>>> parser = PDBParser(PERMISSIVE=1)
>>> parser = PDBParser() # The same (default)
# Strict parser
>>> strict_parser = PDBParser(PERMISSIVE=0)
在宽松状态(默认状态)下,明显包含错误的 PDB 文件将被“纠正”(即,一些残基或原子被排除)。这些错误包括
多个残基具有相同的标识符
多个原子具有相同的标识符(考虑到 altloc 标识符)
这些错误表明 PDB 文件中存在真实问题(有关详细信息,请参阅 Hamelryck 和 Manderick,2003 [Hamelryck2003A])。在严格状态下,包含错误的 PDB 文件将导致异常发生。这有助于查找 PDB 文件中的错误。
但是,一些错误会自动修正。通常,每个无序原子都应具有一个非空白的 altloc 标识符。然而,许多结构不遵循此约定,并且对同一原子的两个无序位置具有一个空白和一个非空白标识符。这将自动以正确的方式解释。
有时,结构包含属于链 A 的残基列表,接着是属于链 B 的残基,然后又是属于链 A 的残基,即链被“打断”。这也被正确地解释了。
示例
PDBParser/Structure 类已在约 800 个结构(每个结构属于一个独特的 SCOP 超家族)上进行了测试。这大约需要 20 分钟,或者平均每个结构 1.5 秒。解析大型核糖体亚基(1FKK)的结构,该结构包含约 64000 个原子,在 1000 MHz PC 上需要 10 秒。
在无法构建明确的数据结构的情况下,生成了三个异常。在这三种情况下,可能的原因是 PDB 文件中的错误,应该加以纠正。在这些情况下生成异常比冒着错误描述结构的风险要好得多。
重复残基
一个结构在一个链中包含两个具有相同序列标识符(resseq 3)和 icode 的氨基酸残基。经过检查发现,此链包含残基 Thr A3、…、Gly A202、Leu A3、Glu A204。很明显,Leu A3 应该是 Leu A203。结构 1FFK 中存在几个类似的情况(例如,包含 Gly B64、Met B65、Glu B65、Thr B67,即残基 Glu B65 应该是 Glu B66)。
重复原子
结构 1EJG 在链 A 的位置 22 处包含 Ser/Pro 点突变。反过来,Ser 22 包含一些无序原子。正如预期的那样,所有属于 Ser 22 的原子都具有非空白的 altloc 说明符(B 或 C)。所有 Pro 22 的原子都具有 altloc A,除了 N 原子,它具有空白的 altloc。这会生成一个异常,因为在点突变处属于两个残基的所有原子都应该具有非空白的 altloc。事实证明,这个原子可能由 Ser 和 Pro 22 共享,因为 Ser 22 缺少 N 原子。同样,这表明文件中存在问题:N 原子应该存在于 Ser 和 Pro 22 中,在这两种情况下都与合适的 altloc 标识符相关联。
自动修正
一些错误非常常见,并且可以很容易地修正,而不会有太多错误解释的风险。这些情况列在下面。
无序原子的空白 altloc
通常,每个无序原子都应具有一个非空白的 altloc 标识符。然而,许多结构不遵循此约定,并且对同一原子的两个无序位置具有一个空白和一个非空白标识符。这将自动以正确的方式解释。
断开的链
有时,结构包含属于链 A 的残基列表,接着是属于链 B 的残基,然后又是属于链 A 的残基,即链被“打断”。这被正确地解释了。
致命错误
有时,无法明确地解释 PDB 文件。与其猜测并冒着错误的风险,不如生成一个异常,并希望用户修正 PDB 文件。这些情况列在下面。
重复残基
链中的所有残基都应该具有唯一的 ID。此 ID 是根据以下内容生成的
序列标识符(resseq)。
插入代码(icode)。
hetfield 字符串(“W” 代表水,“H_” 后跟其他杂原子的残基名称)
点突变情况下残基的残基名称(为了将 Residue 对象存储在 DisorderedResidue 对象中)。
如果这没有导致唯一的 ID,那么很可能存在错误,并且会生成一个异常。
重复原子
残基中的所有原子都应该具有唯一的 ID。此 ID 是根据以下内容生成的
原子名称(不带空格,或者如果出现问题则带空格)。
altloc 说明符。
如果这没有导致唯一的 ID,那么很可能存在错误,并且会生成一个异常。
访问蛋白质数据库
从蛋白质数据库下载结构
可以通过对 PDBList 对象使用 retrieve_pdb_file 方法,从 PDB(蛋白质数据库)下载结构。此方法的参数是结构的 PDB 标识符。
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file("1FAT")
PDBList 类也可以用作命令行工具
python PDBList.py 1fat
下载的文件将被称为 pdb1fat.ent,并存储在当前工作目录中。请注意,retrieve_pdb_file 方法还有一个可选参数 pdir,它指定要存储下载的 PDB 文件的特定目录。
retrieve_pdb_file 方法还有一些选项可以指定用于下载的压缩格式,以及用于本地解压缩的程序(默认 .Z 格式和 gunzip)。此外,在创建 PDBList 对象时,可以指定 PDB ftp 站点。默认情况下,使用全球蛋白质数据库的服务器 (ftp://ftp.wwpdb.org/pub/pdb/data/structures/divided/pdb/)。有关更多详细信息,请参阅 API 文档。再次感谢 Kristian Rother 捐赠此模块。
下载整个 PDB
以下命令将所有 PDB 文件存储在 /data/pdb 目录中
python PDBList.py all /data/pdb
python PDBList.py all /data/pdb -d
此 API 方法被称为 download_entire_pdb。添加 -d 选项将把所有文件存储在同一个目录中。否则,它们将根据其 PDB ID 按 PDB 风格的子目录进行排序。根据流量,完整的下载将需要 2-4 天。
保持本地 PDB 副本更新
这也可以使用 PDBList 对象完成。只需创建一个 PDBList 对象(指定本地 PDB 副本所在的目录)并调用 update_pdb 方法
>>> pl = PDBList(pdb="/data/pdb")
>>> pl.update_pdb()
当然,您可以为此创建一个每周的 cronjob,以使本地副本自动保持最新。也可以指定 PDB FTP 站点(参见 API 文档)。
PDBList 还有一些额外的有用方法。可以使用 get_all_obsolete 方法获取所有已过时的 PDB 条目列表。可以使用 changed_this_week 方法获取本周添加、修改或作废的条目。有关 PDBList 功能的更多信息,请参阅 API 文档。
一般问题
Bio.PDB 的测试程度如何?
实际上,测试相当完善。Bio.PDB 已在近 5500 个 PDB 结构上进行了广泛测试 - 所有结构似乎都已正确解析。更多详细信息可以在 Bio.PDB 生物信息学文章中找到。Bio.PDB 已被许多研究项目用作可靠工具,而且正在被使用。事实上,我几乎每天都将 Bio.PDB 用于研究目的,并继续致力于改进它并添加新功能。
它的速度如何?
在约 800 个结构(每个结构属于一个唯一的 SCOP 超家族)上测试了 PDBParser 的性能。这大约需要 20 分钟,平均每个结构需要 1.5 秒。在一个 1000 MHz 的 PC 上解析大型核糖体亚基(1FKK)的结构(包含约 64000 个原子)需要 10 秒。简而言之:对于许多应用程序来说,它已经足够快了。
是否支持分子图形?
不直接支持,主要是因为已经存在不少基于 Python/支持 Python 的解决方案,这些解决方案可以与 Bio.PDB 结合使用。顺便说一下,我选择使用 Pymol(我已成功将它与 Bio.PDB 结合使用,而且 Bio.PDB 中可能很快就会有/将来会有一些专门的 PyMol 模块)。基于 Python/支持 Python 的分子图形解决方案包括
PyMol:https://pymol.org/
CCP4mg:http://www.ccp4.ac.uk/MG/
谁在使用 Bio.PDB?
Bio.PDB 用于构建 DISEMBL,这是一个预测蛋白质中无序区域的网络服务器 (http://dis.embl.de/)。Bio.PDB 还被用来对 PDB 中蛋白质结构之间的活性位点相似性进行大规模搜索 [Hamelryck2003B],以及开发一种识别线性二级结构元素的新算法 [Majumdar2005]。
从对功能和信息的请求来看,Bio.PDB 也被一些 LPC(大型制药公司 :-)使用。