用于计算分子生物学的 Python 工具
用于计算分子生物学的 Python 工具
ABI 跟踪文件可能有助于检查。虽然有现成的解决方案可用,但您可能会发现自己需要编写自己的代码以以编程方式与跟踪进行交互。Biopython 允许我们这样做。
我在下面展示了一个交互式 IPython 终端会话,使用我自己的 Sanger 测序文件,但它将适用于其他 AB1 文件。
首先,我们读取 ABI 文件
from Bio import SeqIO
record = SeqIO.read("55-Mn-fw-EM-28.ab1", "abi")
我们最感兴趣的数据在记录的注释属性中。
>>> list(record.annotations.keys())
dict_keys(["dye", "abif_raw", "sample_well", "run_finish", "machine_model", "run_start", "polymer"])
在 abif_raw 下是另一个数据字典。
>>> list(record.annotations["abif_raw"].keys())
dict_keys(["DATA5", "DATA8", "RUNT1", "phAR1", ..., "DATA6"])
根据 ABI 数据规范(第 40 页),所有常规显示的跟踪所需的数据都在 DATA9 到 DATA12 通道中。我们可以以编程方式获取这些通道。但是,它并没有明确说明哪个字母对应于哪个颜色,因此哪个通道是确切的。
from collections import defaultdict
channels = ["DATA9", "DATA10", "DATA11", "DATA12"]
trace = defaultdict(list)
for c in channels:
trace[c] = record.annotations["abif_raw"][c]
现在,可以将它们绘制在 matplotlib 图上。
plt.plot(trace["DATA9"], color="blue")
plt.plot(trace["DATA10"], color="red")
plt.plot(trace["DATA11"], color="green")
plt.plot(trace["DATA12"], color="yellow")
plt.show()
放大特定区域后,我们将获得以下轨迹。
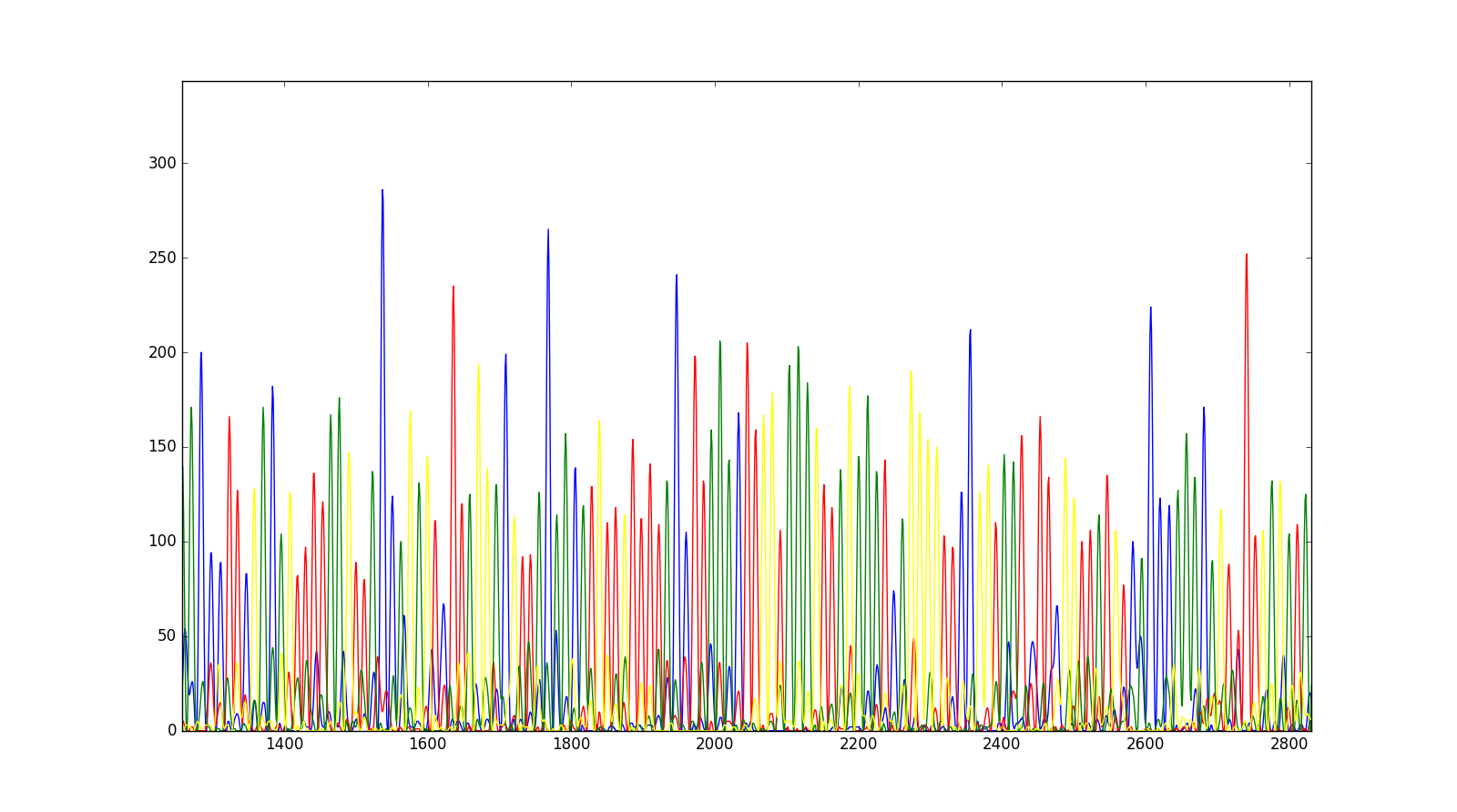
截至目前,此食谱并未进一步深入计算指标。一件可能令人感兴趣的事情是量化混合碱基调用与单碱基调用的香农多样性。
需要注意的另一件事:数据中每个碱基有 10 个色谱图值。因此,这意味着最好每隔 5 个值获取一个,这样序列跟踪数组的最终长度与应该测序的位点数量相匹配,而不是比它大 10 倍。